|
 |
 |
Clarification
of Mechanisms Underlying
Development of Hereditary Parkinsonユs Disease
Laboratory for Motor
System Neurodegeneration |
|
|
Background
of study
Parkinsonユs disease is a neurodegenerative disease, the main symptom of which
is progressive motor disturbance. In Japan, the number of patients with the disease
is estimated to be approximately one hundred thousand. Neuropathologically, the
disease is characterized by selective degeneration of dopamine-producing neurons
in the midbrain nigra. Parkinsonユs disease is believed to be hereditary in approximately
5% of patients. Autosome recessive hereditary juvenile Parkinsonism (AR-JP) occurs
particularly in those who are 40 years or under. The cause of AR-JP is the lack
of the gene called Parkin. In 2000, we and other groups clarified that Parkin
is an enzyme which promotes decomposition of a particular protein. Based on this
finding, we considered that a protein (substrate) that should have been decomposed
by Parkin is accumulated in dopamine-producing neurons, resulting in their degeneration.
We further considered that clarification of the substrate of Parkin is the key
to revealing mechanisms underlying the development of AR-JP. Currently, we, a
team involved in the study of motor neurodegeneration in cooperation with Juntendo
University, succeeded in identifying the substrate of Parkin, which is believed
to play a significant role in the development of AR-JP.
Results
Using the yeast two-hybrid system, the Parkin-associated endothelin receptor-like
(Pael) protein was identified as the membrane protein that binds to Parkin. The
following have also been clarified.
1. Decomposition of Pael is promoted by Parkin.
2. Pael is difficult to fold properly. Pael that fails to fold properly is rapidly
decomposed by Parkin.
3. In experiments using cultured cells, when decomposition of Pael is inhibited
by drugs, unfolded Pael accumulates abnormally in the endoplasmic reticulum (ER).
Finally, cell death occurs due to stress on the ER.
4. Pael is not decomposed in the brain of patients with AR-JP and is accumulated
in amounts 10 to 30 times greater than that in the brains of patients without
AR-JP.
5. Pael is actively expressed in oligodendroglia cells in the brain and in general,
less actively in neurons. However, Pael is particularly expressed in exceptionally
high amounts in dopamine-producing neurons in the midbrain nigra, which has lesions
in AR-JP proteins.
Based on these findings, we assume that in AR-JP, due to abnormal accumulation
of Pael that fails to fold properly, cell death selectively occurs in some dopamine-producing
neurons, resulting in development of AR-JP (refer to Figure).
Prospects for the future
We expect that based on this study a treatment method that targets Pael and prevents
degeneration of dopamine-producing neurons will be developed. In concrete terms,
by inhibiting production of Pael or promoting decomposition of Pael, treatment
for AR-JP will be realized. In addition, this study showed that abnormal accumulation
of proteins, such as Pael, that did not fold properly in neurons may possibly
cause neurodegeneration as in sporadic Parkisonユs disease. Studying this possibility
further may enable us to develop a treatment method for Parkinsonユs disease. |
Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y. and Takahashi, R. An unfolded
putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress,
is a substrate of Parkin. Cell, 105, 891-902, 2001.
| |
|
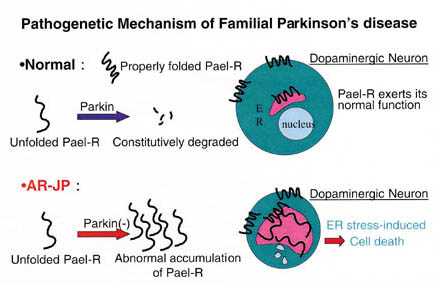 |
| Mechanisms
underlying development of hereditary Parkinsonユs disease: when Parkin failure
occurs, unfolded Pael accumulates abnormally in the ER, causing ER stress and
ultimately cell death. |
|
 |
 |
|
|