|
|
Q
What are the characteristics of CAG repeat diseases, and what are the factors that cause them? |
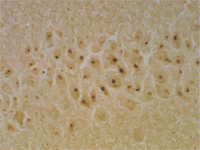
Fig.1
Model Mouse for Huntington's Disease The inclusion body, which is stained by anti-ubiquitin antibody, is seen in the nucleus. This pathological change is also recognized in a lesion in humans.
|
A. CAG repeat diseases currently recognized today include Huntington's disease, known as chorea for its strong involuntary movement, hereditary spinocerebellar ataxia characterized by its cerebellar ataxia, bulbospinal (bulbar and spinal) muscular atrophy known for its damage to motor neurons. CAG repeat diseases are hereditary disorders, bulbospinal muscular atrophy is an X-linked recessive hereditary disease, and all the other diseases are of autosomal dominant heredity.
In a human genome, there are repeats of three nucleotides (triplets) -- CAG, CCG and CTG, and the number of repeats varies from person to person. In the genes of a normal person, this number is restricted to some extent, while an excessive repeat expansion is observed in the genes of patients suffering from a CAG repeat disease and other repeat disease. These disorders are also known as "triplet repeat diseases".
The expanded GAG repeat is located within the coding regions of a responsible gene protein. This suggests that the translated glutamine expansion from CAG repeat plays an important role in the pathomechanisms of these diseases.
|
Q
Why was it that you got started in researching these diseases?
A. I was originally a doctor of neurology, and had examined a large number of patients who suffered from neurodegenerative disorders. They included many hereditary disorders, however, the responsible disease genes were not known. In addition, treatment was so difficult that all we could do was to reduce the symptoms and to encourage the patients.
Over the past ten years, it has become possible to locate abnormal genes related to these diseases. However, we were faced with quite a dilemma, because even when we could identify a disease gene, we were still unclear about the pathomechanisms of that disease. Also, even if we arrived at the correct diagnosis, there was no means of treatment. We were able to diagnose the abnormal genes in healthy individuals (carriers) but could not prevent them from developing the diseases.
I wanted to elucidate the pathomechanisms of these hereditary disorders and to conduct research on critical prevention based on this. CAG repeat diseases are diseases that have common mechanisms, and I believed that the findings on their pathomechanism could be applicable in the treatment of a relatively large number of patients, as well as in preventing their families from suffering from the diseases too. Also, I expected that promoting this research might have a ripple effect on elucidating the pathomechanism of diseases caused by a single abnormal gene (signal gene disorders).
Q
What kinds of research have been conducted in the past?
A. It has been rather difficult to examine disorders such as CAG repeat diseases, in which a neuron gene dies without leaving anything behind. However, positional cloning techniques have broken this barrier. In research on hereditary spinocerebellar ataxia in Japan, the genes that cause DRPLA, MJD and SCA2 have been identified. These achievements are owed to the contributions by neurologists and neuropathologists who have been classifying those diseases clinically and pathologically.
Recently, we have followed the basic approach on other fields and are trying to identify gene products, as well as to make a cellular system, which express a gene product in order to understand their functions. Also, in our research fields, we are producing "knock out" mice and transgenic mice into which abnormal genes are introduced.
Q
What are the positional cloning techniques?
A. Today, there exist numerous genetic markers in various locations of chromosomes. Our analysis of hereditary disorders is based on these markers. Polymorphism is observed in these genetic markers, and we assume that each patient with a disease has the same genetic marker, pattern A, while normal subjects show a different pattern. Then, we consider that the disease gene exists in the vicinity of the genetic marker, pattern A, and proceed with further detailed analysis to finalize the gene.
In this approach, it has become possible to identify the genes of disorders for which we were previously unable to identify disease genes in a biochemical approach. Even so, there still remain many pathomechanisms left that need to be explained.
|
| Q
What kinds of research are you pursuing in your laboratory? |
A. In our laboratory, we are investigating CAG repeat diseases as models of single gene disorders. The research focuses on the production of model systems. We are currently producing both cellular and mice models. For the cellular model, we are attempting to induce cell death by using a model that is expressing GFP (green fluorescent protein) with polygluta-mine. Using a transgenic mice model, we are investigating the differences between the cellular model system and mice model system.
In the cellular model cells die in about four days while forming aggregates, however, in the mouse neurons remains alive even when they contain aggregates. Supposedly, there is a mechanism in the mice system that inhibits cell death, so the cells are able to survive even in a morbid state. We use the differential display analysis in order to search for genes that cause these differences.
Q
How is your research into other research fields of neurological disorders going?
A. Since CAG repeat diseases are caused by protein accumulation like neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease, a common approach of prevention might possibly be discovered in terms of the conformational changes in a protein. Also if the resulting cell deaths share certain similarities, a common inhibitor might be possibly found as well.
|
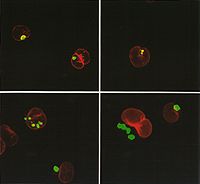
Fig.2
Cellular Model
The genes of Huntington's disease with green fluorescent protein (GFP) were introduced into a mouse's neuroblastoma cell. Protein aggregations are observed in the nucleus and in the cytoplasm.
|
|
magnified scene by clicking image
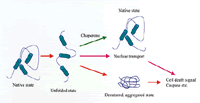
Fig.3
Conformational change of the protein induced neurodegeneration
In a normal condition, proteins take up a stable conformation (native state). However, a protein with mutation takes unstable conformation (unfold condition), and is distributed abnormally (such nuclear transport), or aggregates, which then activate all death signals. The protein called chaperon acts to maintain the normal conformation, and the caspase inhibitor is capable of inhibiting cell death, both suggesting the potential for a cure of these disorders.
|
Q
What about research into cures and the development of new treatment techniques?
A. Unfortunately, there's still a lot left to be done in the development of new treatments. Recently, it has been reported that the disease-developing process in transgenic animals can be inhibited by introducing a caspase inhibitor. Although this will not be an intrinsic method of disease prevention, it could possibly yield new prospects for neurodegenerative disorders that were once considered impossible to cure.
For CAG repeat diseases, there has been considerable progress in research that uses model systems. If some substance effective on relevant models is discovered in the future, it may not be so long until we are able to test it directly on patients. It is difficult to tell when this will actually come about, but I can say that the discouraging outlook we generally had two years ago when I was practicing medicine, where it was felt that it was virtually impossible to prevent neurodegenerative disorders, no longer exists.
Q
Finally, please tell us about your intentions and hopes for your research in the future.
A. My impression is that research progress is ultimately influenced by the number of researchers who conduct it.
About fifteen years ago, there weren't so many researchers working on Alzheimer's disease. However, the number of researchers has been rapidly increasing, due to expansions in research budgets that have resulted from a growing aged population, and the emergence of venture businesses and pharmaceutical enterprises who expect future potential in this field. I believe that this trend has supported the recent rapid progress in research of Alzheimer's disease.
It is currently possible at RIKEN and similar basic research institutions to investigate disorders such as CAG repeat diseases. This will lead to further increases in the number of researchers in the field of neurodegenerative disorders, and in fact, our laboratory is attracting staff with all different kinds of backgrounds.
Even though I no longer examine patients first hand, I believe that we are in an ideal position to actually begin to find ways to prevent these kind of intractable diseases through the combined energies of researchers and through joint research with businesses. Hopefully by doing so, we will be able to help those suffering from these diseases as well as their families.
|
|
|
|
|
 |
|
|