 |
 |
 |
遺伝性パーキンソン病の発症メカニズムを解明
運動系神経変性研究チーム |
|
|
研究の背景
パーキンソン病は進行性の運動障害を主症状とする神経変性疾患で、日本での患者数は約10万人と推定されています。神経病理学的には中脳黒質のドーパミンを産生する神経の選択的変性が特徴です。パーキンソン病のうち、約5%の患者は遺伝性と考えられます。常染色体劣性遺伝性若年性パーキンソニズム(AR-JP)は40才以下で発症する特異なパーキンソン病で、パーキンと呼ばれる遺伝子の欠損が原因です。2000年、我々及び他のグループによってパーキンが特定の蛋白質の分解を促進する酵素であることが示されました。この発見から、AR-JPではパーキンが本来分解すべき蛋白質(基質)がドーパミン神経に蓄積し、神経変性を引き起こすことが予想されました。すなわちパーキンの基質を明らかにすることがAR-JPの発症メカニズム解明への鍵と考えられるに至りました。今回、運動系神経変性研究チームは順天堂大学との共同研究でAR-JPの発症に重要な役割を果たすと考えられるパーキンの基質の同定に成功しました。
成果
酵母two-hybrid法でパーキン結合蛋白として膜蛋白質であるパエル受容体(Pael:Parkin associated endothelin receptor-like)が同定され、以下のことが明らかになりました。
1. パエル受容体はパーキンによって分解が促進される。
2. パエル受容体は正常な形に折れたたむこと(フォールディング)が難しいタンパク質であり、フォールディングに失敗したパエル受容体は、パーキンの作用ですみやかに分解される。
3. 培養細胞での実験では、薬剤によって分解を抑制すると、フォールディング不全のパエル受容体が小胞体に異常蓄積し、細胞は小胞体ストレスによる細胞死に陥る。
4. パエル受容体は、AR-JPの患者脳で分解されずに、非AR-JP対照脳の10〜30倍以上蓄積している。
5. パエル受容体は、脳のオリゴデンドログリア細胞に強く発現し、一般的には神経細胞での発現は乏しいが、AR-JPの病変部位である中脳黒質のドーパミン神経では例外的に強く発現している。
これらの事実から、AR-JPではフォールディングに失敗したパエル受容体の異常蓄積により、ドーパミン神経が選択的に細胞死に陥って発症に至るのではないかと考えられます(図)。
今後への期待
本研究により、パエル受容体をターゲットとして、AR-JPでのドーパミン神経の変性を防ぐ治療法が生み出されることが期待されます。具体的にはパエル受容体の産生を抑えたり、また分解を促進することでAR-JPの治療が可能になるでしょう。また本研究によって普通のパーキンソン病においても、パエル受容体のようにフォールディングが困難な蛋白質の神経細胞への異常蓄積が神経変性の原因になっている可能性が新たに提示されました。この可能性を追究することで、まったく新しい観点からパーキンソン病の根治療法が生まれる可能性があります。 |
Imai,
Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y. and Takahashi, R. An unfolded
putative transmembrane polypepyide, which can lead to endoplasmic reticulum stress,
is a substrate of Parkin. Cell, 105, 891-902, 2001
| |
|
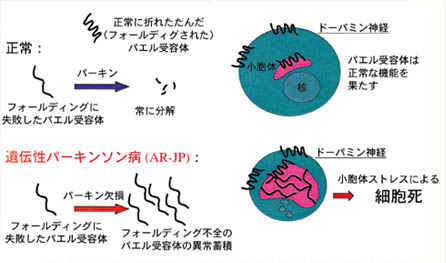
図 遺伝性パーキンソン病の発症メカニズム
パーキンが欠損すると、フォールディング不全のパエル受容体が小胞体に異常蓄積し、細胞死を引き起こす。
|
|
 |
 |
|
|