




Alzheimer's disease (AD) is a progressive form of dementia and, as the risk of AD increases with age, a major concern for aging societies. The only approved drug to treat AD in Japan is Aricept (donepezil hydrochloride). This drug inhibits the enzyme that degrades acetylcholine, a neuro-transmitter that is reduced in AD brains. The limited effectiveness of this drug makes it suitable only as a palliative treatment. Therefore, another approach for therapy would be to target other processes associated with the pathological aspects of AD, such as the accumulation of excess amyloid peptide, Aβ, which is associated with AD. We assume that AD may be treated or prevented by reducing, eliminating, or even preventing the accumulation of Aβ. In this vein, we first identified an enzyme that can degrade Aβ in the brain, neprilysin and found that aging reduces the expression of neprilysin. Reduced expression of the enzyme is also seen just prior to AD onset. After identifying the activity of neprilysin, we noticed that accelerated Aβ accumulation is correlated with reduced neprilysin activity that we confirmed using genetic manipulation to increase neprilysin activity resulting in reduced Aβ in the brain. It is possible that selectively increasing neprilysin expression might delay or prevent the onset of AD.
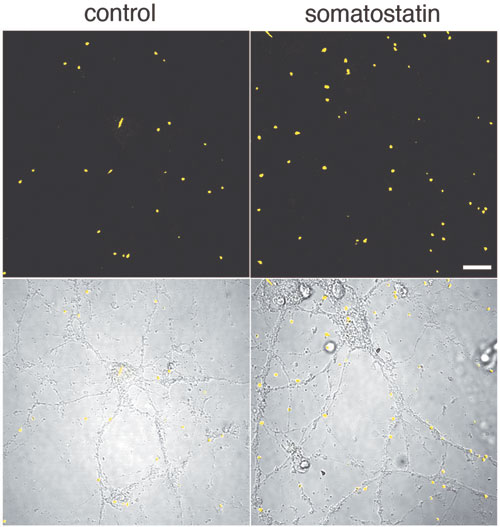
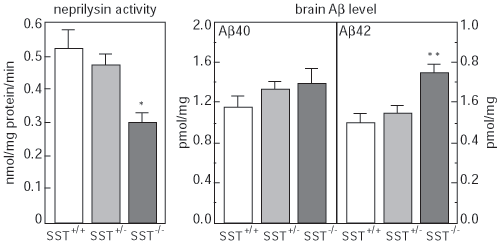
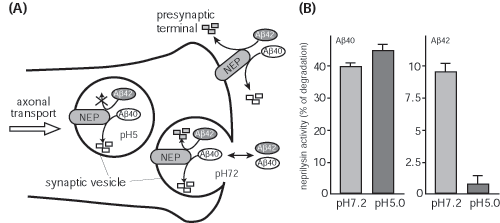
To test this, we first developed a method to estimate neprilysin activity and screened for those factors that increase neprilysin activity. We found that somatostatin (SST), a neuropeptide, accelerates this activity (Fig. 1). We know that, like acetylcholine, levels of SST decrease in AD brain; however, the reason for this is unknown. Taking these in vitro screening results into consideration, we examined how SST might control the activity of neprilysin and establish an SST-KO mouse. We found that of factors known to participate in Aβ metabolism only neprilysin expression was associated with SST. Neprilysin activity in the hippocampus of SST-KO mice was about 60% less than that of wild-type. Aβ levels increased in the brain when a counter measure was applied to the mouse. Significant and selective increases in the level of Aβ42, which has a higher degree of neurotoxicity than Aβ40, were observed (Fig. 2). Further analysis revealed a decrease in expression and an abnormal pattern of localization for neprilysin in the SST-KO mouse. Neprilysin, localizes at the presynaptic terminal, however, the neprilysin localization in the mutant mouse was lower at that terminal. This abnormality suggests that neprilysin can only degrade Aβ42 at the presynaptic terminal (Fig. 3). Neprilysin can degrade Aβ40 at both intercellularly and at the presynaptic terminal. Hence, SST selectively activates the degradation of Aβ42 by controlling the expression and localization of neprilysin.
Five different somatostatin receptors (SSTR) identified in SST. Isolating SSTR regulation of neprilysin activity, may become a drug that selectively targets the appropriate SSTR to help reduce Aβ levels in brain, efficiently and with minimal side effects. As the SST level decreases with normal aging and in AD, any drug exploiting this process would be useful as a replacement therapy. Aβ levels in the brain may be lowered if neprilysin activity is reved up by a factor of 2. Down the road, a drug that targets this SSTR and that could efficiently decrease Aβ levels in the brain, might also be able to prevent AD by causing slight increases in neprilysin activity. Our results suggest that therapeutic prevention for AD may be possible using a new approach and contribute to our understanding of the mechanisms of onset in most AD patients. Further developments from this research are anticipated.
