




Background
Certain neurological diseases, such as Huntington disease or hereditary spinocerebellar ataxias, are characterized by excessive repetitions of CAG-base sequences in the diseasecausing gene. The products of these repeat sequences, including abnormally expanded glutamine chains, accumulate in neurons eventually causing cell death and functional abnormalities. These diseases are collectively referred to as polyglutamine diseases because of the presence of abnormal glutamine chains. The longer the polyglutamine change, the earlier disease onset. Therefore, most research on the pathology of these diseases has focused on relationship between the length of the polyglutamine chain and neuronal toxicity. Recent analyses of mouse models of these diseases have shown that protein aggregates containing polyglutamine form inclusion bodies in the nuclei of affected neurons. This suggests that aggregate formation might play an important role in the progress of these diseases. Yet, even as the pathology of polyglutamine diseases is slowly revealed, suitable therapies to treat them have yet to be established. Gene diagnosis has not led to treatment. Thus, our focus is to understand the pathology so as to establish a suitable therapy and prevention methods for these diseases.
Outline of Research
The Laboratory for Structural Neuropathology at RIKEN Brain Science Institute clarified the mechanisms of these diseases by studying the developing polyglutamine in cell models. We found that elongated polyglutamine chains develop and aggregate in cells. When the chaperone system that should remove this aggregation fails and proteolysis is inhibited, aggregates accumulate and cell death occurs. Precisely how these aggregates form and recruit various molecules lies at the core of the pathology of these diseases.
It is possible that controlling aggregation might be the means to regulate progression of the diseases.Therefore, we analyzed structural abnormalities present in those proteins, including polyglutamine, that were located upstream of the pathology. We inserted various polyglutamine chains into myoglobin, which is an extremely stable protein with a known structure, and examined the structure of both the polyglutamine chain and the resulting structural changes of the host protein. Longer polyglutamine chains take an intramolecular beta sheet structure. Mutant myoglobin between 35 and 50 repeats had intermolecular beta sheet structures at the start of aggregate formation. These myoglobin proteins also destabilized as the polyglutamine chain expanded. Using this mutant myoglobin system, we searched for compounds that could inhibit aggregate formation.
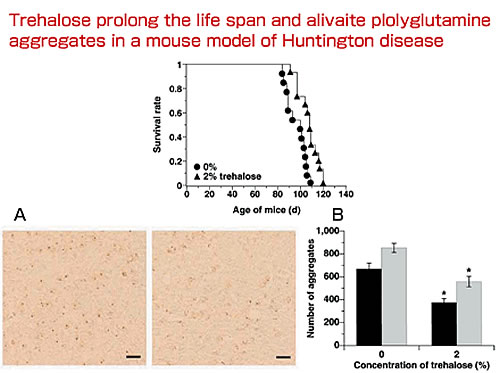
We analyzed more than 200 compounds in vitro using Mb-Q35 (myoglobin containing the 35 polyglutamine) and found that disaccharides, trehalose in particular, mediated aggregate formation. We then confirmed the role of trehalose in regulating aggregation in affected neurons. Trehalose-synthesizing enzymes Ots A and B were expressed in neurons. We observed inhibiting effects on aggregation formation were similar to those obtained with chaperone HDJ1. We also found that Mb-Q35 in the presence of trehalose is as stable as Q12 (myoglobin containing 12 glutamine repeats). Trehalose appears to be able to stabilize molecules and inhibit aggregation, thereby reducing cell death. In addition, when a 2% concentration of trehalose was administered to R6/2 Huntington disease model mice through their drinking water, death was delayed, they survived 10% longer than expected. In addition, functional deterioration, tested by rotarod, was delayed and aggregate formation was lower than in untreated mice. Trehalose appears to inhibit the destabilization of host proteins caused by expanded polyglutamine and aggregate formation and delay the progress of polyglutamine diseases.
The Significance of the Research
We have shown that aggregates formation occurs at the upstream of polyglutamine diseases pathogenesis and that reducing this aggregation can delay the progress of the disease. However, this is not a perfect remedy. Searching for other compounds that can exert more effective control over the accumulation and degradation of aggregates is required. Success here may yield suitable therapies in the future. Misfolded protein is a common characteristic in many neurodegenerative disorders, including Alzheimer’s and Parkinson’ s diseases and amyotrophic lateral sclerosis. We believe that our results on molecular stabilization might also point to potential avenues to therapies for these diseases.
