




Our laboratory has discovered a nonsense mutation (a mutation that causes the premature termination of transcription to produce an incomplete and nonfunctional amino acid) of SCN2A gene, that codes the voltage-gated sodium channel-subunit type 2, in a patient with intractable epilepsy and severe mental impairment. The protein truncated by this mutation altered the functions of the remaining normal-channel proteins. We previously found a missense mutation (that is a mutation which causes the amino acid substitution) in the SCN2A gene in a patient with milder epilepsy and showing no mental decline. Different mutations in the same gene appear to produce differences in the severity of the epilepsy and in the dominant-negative effect of the truncated channel protein. These findings suggest a new disease-causing mechanism, contribute to the understanding of the molecular pathology of epilepsy, and may lead to the development of effective therapies for this disease.
Background
Epilepsy is a neurological disorder which affects more than 1% of the population. It is characterized by repeated attacks of motor, sensory, or psychic function that can include tonic-clonic seizures, myoclonia, and loss of consciousness. There are many types of epilepsy most of which are considered to have a genetic background, more than 30 etiogenic genes have been identified in association with the disease. Epilepsy can be roughly categorized as either idiopathic epilepsy, a relatively mild form with only the occurrence of seizures, or symptomatic epilepsy, a more severe and intractable form of epilepsy that is accompanied by ataxia and/or mental decline. Most of the 16 genes identified as causing idiopathic epilepsy are known to code ion channels. Voltage-gated sodium channels are the primary factors for neuron excitation. Mutations of SCN1A and SCN2A genes which code for the sodium channel-subunit type 1 protein and sodium channel b1 subunit protein respectively have been reported in generalized epilepsy with febrile seizures plus (GEFS+), an idiopathic epilepsy. In 2001 our laboratory reported on a missense mutation of the SCN2A gene in patients with an idiopathic epilepsy, similar to febrile seizures plus. Later, missense mutations of SCN2A were also found in cases of Benign Familial Neonatal Convulsion (BFNC), an even milder form of epilepsy. Recently, mutations of SCN1A were identified in a severe form of epilepsy: Severe Myoclonic Epilepsy in Infancy (SMEI). Febrile convulsions first appear in the first year of life and are followed by intractable tonic clonic seizures, myoclonia and severe mental retardation. Our laboratory has also identified SCN1A mutations in Intractable Childhood Epilepsy with Generalized Tonic-Clonic (ICEGTC), which is a kind of SMEI that is not accompanied by myoclonia. Although these mutations occur in the same SCN1A gene, those observed in the patients with GEFS+ are almost always missense mutations, while nearly two-thirds of those observed in the patients with SMEI are either nonsense or frame-shift mutations.
Results
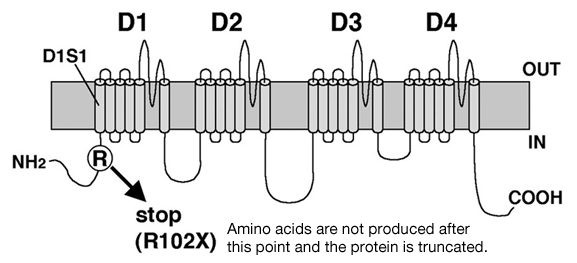
We took DNA samples from the blood of 60 patients diagnosed as having SMEI or an atypical related case. Mutational analyses were conducted on SCN1A and SCN1B genes by reading DNA base sequences. In 20 samples showing no mutations in SCN1A or SCN1B genes, the SCN2A genes were also analyzed. In one of these samples, a c. 304C>T (R102X) mutation was found. In this mutation the 304th base in the cDNA was altered from C (cytosine) to T (thymin), changing the codon for R (arginine), which is the 102nd amino acid in the channel protein, to X (the stop codon)) of the SCN2A gene (Fig.). Therefore, protein synthesis is halted not far from the NH2 amino terminal. This mutation was previously identified as a heterozygous mutation (a mutation in only one of the two genes inherited from parents), in this case it was a new mutation because neither parent showed the same mutation.
The patient’s symptoms were similar to SMEI in that intractable epilepsy and severe mental retardation was apparent, however there were noticeable differences. First, SMEI is normally manifests as a generalized epilepsy, yet the patient with SCN2A-R102X had a partial epilepsy. Second, SMEI generally appears between 2 to 6 months after birth, while this patient showed symptoms at 1 year 7 months. Finally, the heat sensitivity associated with SMEI (leading to crises during bathing and other occasions involving heat) was not present in this patient. To further investigate the effects of this mutation, we expressed wild-type and mutant human SCN2A-cDNA in human cultured cells to examine the functional changes in electrophysiological properties of the channels using patch-clamp analysis. As a result, only the mutation-type channel did not conduct the current as it had been expected. To our surprise, when channel proteins were expressed simultaneously in both wild-type and mutant channels, the current flow in the wild-type channel exhibited an atypical change. As the truncated mutant protein affected the function of the wild-type channel protein, simply halving the channel proteins seems insufficient to cause this patient’s symptoms. A different mechanism, such as the influence of truncated proteins on the remaining normal channel, should also be involved. Nav1.1 channel proteins are localized in neuron bodies while Nav1.2channel proteins are localized mainly in axons and their respective distributions in the brain are different. The types of mutations in SCN1A that are observed in vastly different types of epilepsies with variation in degrees of severity are also observed in SCN2A. This suggests that a common mechanism exists for the two genes. Finding this mechanism would be useful to understanding the overall pathology of these epilepsies.
Future possibilities
Finding a SCN2A gene mutation in an intractable epilepsy accompanied with severe mental decline that involves a truncated protein which alters the function of normal-channel protein will significantly contribute to diagnoses of epilepsies and the understanding of the disease-causing mechanism. These findings might also lead to new therapies for these intractable epilepsies.
