




The Laboratory for Neurogenetics generated disease-model mice where a mutation was introduced into a particular sodium-channel gene, which is found to have a mutation in cases of intractable epilepsy accompanied by a severe intellectual disorder, and ascertained that those mice were found to have severe epileptic seizures and functional abnormalities of inhibitory nerve cells. Furthermore, the laboratory found that this channel protein is originally expressed predominantly at the axon, etc., of a particular type of inhibitory nerve cell, but hardly exists in excitatory nerve cells. This discovery disproves the commonly accepted, conventional theory regarding the distribution of this particular channel protein and will perhaps make a great contribution to understanding the mechanism by which epilepsy develops and thus contribute to the development of therapies for it.
Background
Epilepsy is a neural disease that characteristically involves seizures induced by supranormal excitation of nerve cells, with a high incidence of 1% of the total population. Epilepsy includes many types, a majority of which are assumed to be associated with genetic causes. Multiple causal genes for epilepsy have, in fact, been identified. One of those genes is SCN1A, a gene encoding the voltage-dependent sodium-channel alpha subunit 1 protein. Reports have revealed that 200 disease mutations in this gene have been found in multiple types of epilepsy of differing degrees of severity. It is now well established that SCN1A is the most representative causal gene for epilepsy of those which have been reported. Severe myoclonic epilepsy in infancy (SMEI), one of the types of epilepsy where mutations in SCN1A have been found, is characterized by febrile seizures as the initial symptom in infancy, intractable tonic-clonic seizures and myoclonic seizures, and mental development disorders. SCN1A mutations are found in a surprising 80% of SMEI cases. About two-thirds of such mutations are nonsense or frameshift mutations that introduce premature terminations into the channel protein; and about one-third are missense mutations (i.e., where encoded amino acids change into different amino acids). Voltage-dependent sodium channels are known to be proteins have a principal role in nerve cell excitation; however, their functions (especially, the individual function of each subtype) are still largely unknown. It is therefore important to more fully analyze the original functions of Nav1.1, which is encoded by SCN1A, and elucidate the mechanism of development of epilepsy caused by its mutations in order to understand this disease and develop effective therapies for it.

Results of this study
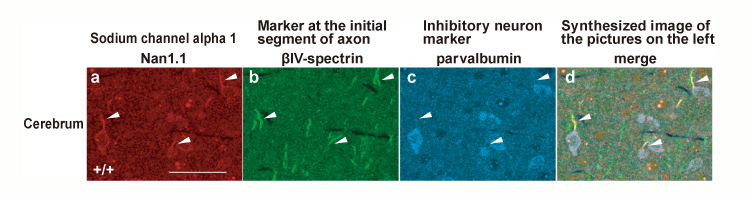
The laboratory produced mice possessing SCN1A with nonsense mutations (R1407X) found in three independent SMEI cases and analyzed them in detail. It was found that the mice with mutations introduced lost the Nav1.1 protein, developed epilepsy, and had inhibitory nerve cell dysfunction. Furthermore, from strictly designed and implemented experiments using multiple antibodies and the mutant mouse as a negative control, it was found that wild mice had Nav1.1 protein expression in particular (parvalbumin-positive) inhibitory nerve cells where a calcium-binding protein called parvalbumin was expressed, and the Nav1.1 expression was especially predominant in their axons. The Nav1.1 protein has traditionally been thought to express itself in the dendrites and somas of both excitable and inhibitory nerve cells. Our discovery, however, that the expression of the protein is localized to the axons of parvalubumin-positive inhibitory nerve cells, etc., disproves this assumption found in prior relevant reports, and it furthermore strongly suggests that the cause of development of epilepsy of the types induced by SCN1A gene mutations, such as SMEI, resides in the weakened functions of parvalubumin-positive cells failing to inhibit the activities of excitable nerve cells.
Expectations
There is high expectation that a means of developing new therapies for severe, debilitating epileptic diseases that are difficult to cure at present will be found by targeting parvalubumin-positive inhibitory nerve cells.
